Genetic Diseases
PDGFRB is the human gene encoding platelet-derived growth factor receptor β (PDGFRβ). Gain-of-function mutations in PDGFRB occur in humans with Penttinen progeroid syndrome and Kosaki overgrowth syndrome. Both of these diseases affect connective tissues, but in opposite ways. Whereas Penttinen syndrome is a progeroid disease with symptoms of accelerated aging, Kosaki syndrome is characterized by increased somatic growth. It is a mystery how different gain-of-function mutations in PDGFRβ could lead to such distinct outcomes for patients.
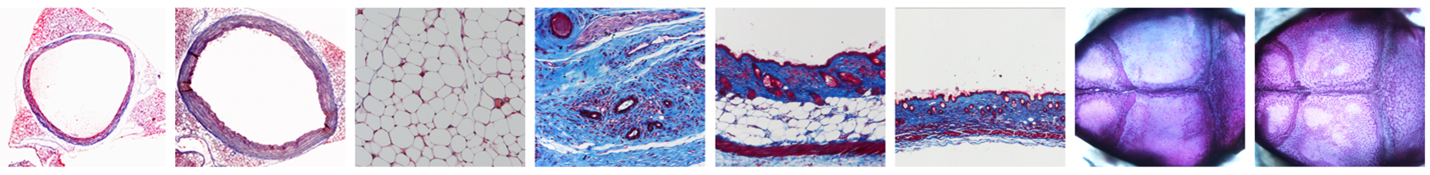
To learn more about PDGFRβ signaling in connective tissue, we generated an allelic series of conditional knockin mice with gain-of-function mutations in Pdgfrb. The first mutation, known as PDGFRβ-D849V, causes hyperactivation of STAT1 and leads to an interferonopathy-like state with connective tissue wasting and vascular inflammation (Olson & Soriano 2011, He et. al., 2015). In terms of signaling, PDGFRβ-D849V phosphorylates STAT1, and activated STAT1 exerts negative feedback on PDGFRβ-D849V signaling. Although STAT1 is best known as a transducer of interferon signaling, our studies show that PDGFRβ-D849V hijacks STAT1 to cause connective tissue wasting/aging. Remarkably, deletion of the Stat1 gene extends the life of PDGFRβ-D849V mice and causes them to become massively overgrown (He et. al., 2017, Kwon et. al., 2021). It is remarkable that Stat1 leads to such different phenotypes: PDGFRβ-D849V mice with Stat1 exhibit premature aging with connective tissue wasting, but PDGFRβ-D849V mice without Stat1 exhibit somatic overgrowth.
We have more recently generated conditional knockin PDGFRβ-V665A and PDGFRβ-P584R mice, bearing mutations respectively found in Penttinen progeroid syndrome and Kosaki overgrowth syndrome. So far, we have determined that PDGFRβ-V665A mice have premature aging of skin, skeleton, and vasculature, and PDGFRβ-P584R mice have somatic overgrowth, thus recapitulating some of the main features of the human diseases. We are in the process of determining how STAT1 and other PDGF signaling proteins are involved with these mutations to generate their distinct phenotypes.